
PUMPED
New member
[h=1]Pharmacokinetics[/h]From Wikipedia, the free encyclopedia

Graph that demonstrates the Michaelis-Menten kinetics model for the relationship between an enzyme and a substrate: one of the parameters studies in pharmacokinetics, where the substrate is a pharmaceutical drug.
Pharmacokinetics, sometimes abbreviated as PK (from Ancient Greek pharmakon "drug" and kinetikos "moving, putting in motion"; see chemical kinetics), is a branch of pharmacology dedicated to determining the fate of substances administered externally to a living organism. The substances of interest include pharmaceutical agents, hormones, nutrients, and toxins. It attempts to discover the fate of a drug from the moment that it is administered up to the point at which it is completely eliminated from the body. Pharmacokinetics describes how the body affects a specific drug after administration through the mechanisms of absorption and distribution, as well as the chemical changes of the substance in the body (e.g. by metabolic enzymes such as cytochrome P450 or glucuronosyltransferase enzymes), and the effects and routes of excretion of the metabolites of the drug.[SUP][1][/SUP] Pharmacokinetic properties of drugs may be affected by elements such as the site of administration and the dose of administered drug. These may affect the absorption rate.[SUP][2][/SUP] Pharmacokinetics is often studied in conjunction with pharmacodynamics, the study of a drug's pharmacological effect on the body.
A number of different models have been developed in order to simplify conceptualization of the many processes that take place in the interaction between an organism and a drug. One of these models, the multi-compartment model, gives the best approximation to reality; however, the complexity involved in using this type of model means that monocompartmental models and above all two compartmental models are the most-frequently used. The various compartments that the model is divided into are commonly referred to as the ADME scheme (also referred to as LADME if liberation is included as a separate step from absorption):
All these concepts can be represented through mathematical formulas that have a corresponding graphical representation. The use of these models allows an understanding of the characteristics of a molecule, as well as how a particular drug will behave given information regarding some of its basic characteristics. Such as its acid dissociation constant (pKa), bioavailability and solubility, absorption capacity and distribution in the organism.
The model outputs for a drug can be used in industry (for example, in calculating bioequivalence when designing generic drugs) or in the clinical application of pharmacokinetic concepts. Clinical pharmacokinetics provides many performance guidelines for effective and efficient use of drugs for human-health professionals and in veterinary medicine.
[h=2]Metrics[edit][/h]The following are the most commonly measured pharmacokinetic metrics:[SUP][5][/SUP]
<tbody>
</tbody>In pharmacokinetics, steady state refers to the situation where the overall intake of a drug is fairly in dynamic equilibrium with its elimination. In practice, it is generally considered that steady state is reached when a time of 4 to 5 times the half-life for a drug after regular dosing is started.
The following graph depicts a typical time course of drug plasma concentration and illustrates main pharmacokinetic metrics:

The time course of drug plasma concentrations over 96 hours following oral administrations every 24 hours. Note that the AUC in steady state equals AUC[SUB]∞[/SUB] after the first dose.
[h=2]Pharmacokinetic models[edit][/h]Pharmacokinetic modelling is performed by noncompartmental or compartmental methods. Noncompartmental methods estimate the exposure to a drug by estimating the area under the curve of a concentration-time graph. Compartmental methods estimate the concentration-time graph using kinetic models. Noncompartmental methods are often more versatile in that they do not assume any specific compartmental model and produce accurate results also acceptable for bioequivalence studies. The final outcome of the transformations that a drug undergoes in an organism and the rules that determine this fate depend on a number of interrelated factors. A number of functional models have been developed in order to simplify the study of pharmacokinetics. These models are based on a consideration of an organism as a number of related compartments. The simplest idea is to think of an organism as only one homogenous compartment. This monocompartimental model presupposes that blood plasma concentrations of the drug are a true reflection of the drug’s concentration in other fluids or tissues and that the elimination of the drug is directly proportional to the drug’s concentration in the organism (first order kinetics).

Graph representing the monocompartmental action model.
However, these models do not always truly reflect the real situation within an organism. For example, not all body tissues have the same blood supply, so the distribution of the drug will be slower in these tissues than in others with a better blood supply. In addition, there are some tissues (such as the brain tissue) that present a real barrier to the distribution of drugs, they can be breached with greater or lesser ease depending on the drug’s characteristics. If these relative conditions for the different tissue types are considered along with the rate of elimination, the organism can be considered to be acting like two compartments: one that we can call thecentral compartment that has a more rapid distribution, comprising organs and systems with a well-developed blood supply; and a peripheral compartment made up of organs with a lower blood flow. Other tissues, such as the brain, can occupy a variable position depending on a drug’s ability to cross the barrier that separates the organ from the blood supply.
This two compartment model will vary depending on which compartment elimination occurs in. The most common situation is that elimination occurs in the central compartment as the liver and kidneys are organs with a good blood supply. However, in some situations it may be that elimination occurs in the peripheral compartment or even in both. This can mean that there are three possible variations in the two compartment model, which still do not cover all possibilities.[SUP][6][/SUP]
This model may not be applicable in situations where some of the enzymes responsible for metabolizing the drug become saturated, or where an active elimination mechanism is present that is independent of the drug's plasma concentration. In the real world each tissue will have its own distribution characteristics and none of them will be strictly linear. If we label the drug’s volume of distribution within the organism Vd[SUB]F[/SUB] and its volume of distribution in a tissue Vd[SUB]T[/SUB] the former will be described by an equation that takes into account all the tissues that act in different ways, that is:
<center style="color: rgb(37, 37, 37); font-family: sans-serif; font-size: 14px; line-height: 22.4px;">
 </center>This represents the multi-compartment model with a number of curves that express complicated equations in order to obtain an overall curve. A number of computer programmes have been developed to plot these equations.[SUP][6][/SUP] However complicated and precise this model may be it still does not truly represent reality despite the effort involved in obtaining various distribution values for a drug. This is because the concept of distribution volume is a relative concept that is not a true reflection of reality. The choice of model therefore comes down to deciding which one offers the lowest margin of error for the type of drug involved.
</center>This represents the multi-compartment model with a number of curves that express complicated equations in order to obtain an overall curve. A number of computer programmes have been developed to plot these equations.[SUP][6][/SUP] However complicated and precise this model may be it still does not truly represent reality despite the effort involved in obtaining various distribution values for a drug. This is because the concept of distribution volume is a relative concept that is not a true reflection of reality. The choice of model therefore comes down to deciding which one offers the lowest margin of error for the type of drug involved.
[h=3]Noncompartmental analysis[edit][/h]Noncompartmental PK analysis is highly dependent on estimation of total drug exposure. Total drug exposure is most often estimated by area under the curve (AUC) methods, with the trapezoidal rule (numerical integration) the most common method. Due to the dependence on the length of 'x' in the trapezoidal rule, the area estimation is highly dependent on the blood/plasma sampling schedule. That is, the closer time points are, the closer the trapezoids reflect the actual shape of the concentration-time curve.
[h=3]Compartmental analysis[edit][/h]Compartmental PK analysis uses kinetic models to describe and predict the concentration-time curve. PK compartmental models are often similar to kinetic models used in other scientific disciplines such as chemical kinetics and thermodynamics. The advantage of compartmental over some noncompartmental analyses is the ability to predict the concentration at any time. The disadvantage is the difficulty in developing and validating the proper model. Compartment-free modelling based on curve stripping does not suffer this limitation. The simplest PK compartmental model is the one-compartmental PK model with IV bolus administration and first-order elimination. The most complex PK models (called PBPK models) rely on the use of physiological information to ease development and validation.
[h=3]Single-compartment model[edit][/h]Linear pharmacokinetics is so-called because the graph of the relationship between the various factors involved (dose, blood plasma concentrations, elimination, etc.) gives a straight line or an approximation to one. For drugs to be effective they need to be able to move rapidly from blood plasma to other body fluids and tissues.
The change in concentration over time can be expressed as C=C[SUB]initial[/SUB]*E^(-kelt)
[h=3]Multi-compartmental models[edit][/h]
Graphs for absorption and elimination for a non-linear pharmacokinetic model.
The graph for the non-linear relationship between the various factors is represented by a curve, the relationships between the factors can then be found by calculating the dimensions of different areas under the curve. The models used in 'non linear pharmacokinetics are largely based on Michaelis-Menten kinetics. A reaction’s factors of non linearity include the following:
[h=2]Bioavailability[edit][/h]Main article: Bioavailability
At a practical level, a drug’s bioavailability can be defined as the proportion of the drug that reaches its site of action. From this perspective the intravenous administration of a drug provides the greatest possible bioavailability, and this method is considered to yield a bioavailability of 1 (or 100%). Bioavailability of other delivery methods is compared with that of intravenous injection («absolute bioavailability») or to a standard value related to other delivery methods in a particular study («relative bioavailability»).
<center style="color: rgb(37, 37, 37); font-family: sans-serif; font-size: 14px; line-height: 22.4px;">
 </center><center style="color: rgb(37, 37, 37); font-family: sans-serif; font-size: 14px; line-height: 22.4px;">
</center><center style="color: rgb(37, 37, 37); font-family: sans-serif; font-size: 14px; line-height: 22.4px;">
 </center>Once a drug’s bioavailability has been established it is possible to calculate the changes that need to be made to its dosage in order to reach the required blood plasma levels. Bioavailability is therefore a mathematical factor for each individual drug that influences the administered dose. It is possible to calculate the amount of a drug in the blood plasma that has a real potential to bring about its effect using the formula:
</center>Once a drug’s bioavailability has been established it is possible to calculate the changes that need to be made to its dosage in order to reach the required blood plasma levels. Bioavailability is therefore a mathematical factor for each individual drug that influences the administered dose. It is possible to calculate the amount of a drug in the blood plasma that has a real potential to bring about its effect using the formula:
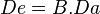 ; where De is the effective dose, B bioavailability and Da the administered dose.
; where De is the effective dose, B bioavailability and Da the administered dose.
Therefore, if a drug has a bioavailability of 0.8 (or 80%) and it is administered in a dose of 100 mg, the equation will demonstrate the following:
<center style="color: rgb(37, 37, 37); font-family: sans-serif; font-size: 14px; line-height: 22.4px;">De = 0.8 x 100 mg = 80 mg</center>That is the 100 mg administered represents a blood plasma concentration of 80 mg that has the capacity to have a pharmaceutical eff
This concept depends on a series of factors inherent to each drug, such as:[SUP][9][/SUP]

Graph that demonstrates the Michaelis-Menten kinetics model for the relationship between an enzyme and a substrate: one of the parameters studies in pharmacokinetics, where the substrate is a pharmaceutical drug.
Pharmacokinetics, sometimes abbreviated as PK (from Ancient Greek pharmakon "drug" and kinetikos "moving, putting in motion"; see chemical kinetics), is a branch of pharmacology dedicated to determining the fate of substances administered externally to a living organism. The substances of interest include pharmaceutical agents, hormones, nutrients, and toxins. It attempts to discover the fate of a drug from the moment that it is administered up to the point at which it is completely eliminated from the body. Pharmacokinetics describes how the body affects a specific drug after administration through the mechanisms of absorption and distribution, as well as the chemical changes of the substance in the body (e.g. by metabolic enzymes such as cytochrome P450 or glucuronosyltransferase enzymes), and the effects and routes of excretion of the metabolites of the drug.[SUP][1][/SUP] Pharmacokinetic properties of drugs may be affected by elements such as the site of administration and the dose of administered drug. These may affect the absorption rate.[SUP][2][/SUP] Pharmacokinetics is often studied in conjunction with pharmacodynamics, the study of a drug's pharmacological effect on the body.
A number of different models have been developed in order to simplify conceptualization of the many processes that take place in the interaction between an organism and a drug. One of these models, the multi-compartment model, gives the best approximation to reality; however, the complexity involved in using this type of model means that monocompartmental models and above all two compartmental models are the most-frequently used. The various compartments that the model is divided into are commonly referred to as the ADME scheme (also referred to as LADME if liberation is included as a separate step from absorption):
- Liberation - the process of release of a drug from the pharmaceutical formulation.[SUP][3][/SUP][SUP][4][/SUP] See also IVIVC.
- Absorption - the process of a substance entering the blood circulation.
- Distribution - the dispersion or dissemination of substances throughout the fluids and tissues of the body.
- Metabolization (or biotransformation, or inactivation) – the recognition by the organism that a foreign substance is present and the irreversible transformation of parent compounds into daughter metabolites.
- Excretion - the removal of the substances from the body. In rare cases, some drugs irreversibly accumulate in body tissue.
All these concepts can be represented through mathematical formulas that have a corresponding graphical representation. The use of these models allows an understanding of the characteristics of a molecule, as well as how a particular drug will behave given information regarding some of its basic characteristics. Such as its acid dissociation constant (pKa), bioavailability and solubility, absorption capacity and distribution in the organism.
The model outputs for a drug can be used in industry (for example, in calculating bioequivalence when designing generic drugs) or in the clinical application of pharmacokinetic concepts. Clinical pharmacokinetics provides many performance guidelines for effective and efficient use of drugs for human-health professionals and in veterinary medicine.
[h=2]Contents[/h] [hide]
[h=2]Metrics[edit][/h]The following are the most commonly measured pharmacokinetic metrics:[SUP][5][/SUP]
| Characteristic | Description | Example value | Symbol | Formula |
|---|---|---|---|---|
| Dose | Amount of drug administered. | 500 mg |
| Design parameter |
| Dosing interval | Time between drug dose administrations. | 24 h | 
| Design parameter |
| C[SUB]max[/SUB] | The peak plasma concentration of a drug after administration. | 60.9 mg/L | 
| Direct measurement |
| t[SUB]max[/SUB] | Time to reach C[SUB]max[/SUB]. | 3.9 h | 
| Direct measurement |
| C[SUB]min[/SUB] | The lowest (trough) concentration that a drug reaches before the next dose is administered. | 27.7 mg/L | 
| Direct measurement |
| Volume of distribution | The apparent volume in which a drug is distributed (i.e., the parameter relating drug concentration to drug amount in the body). | 6.0 L | 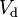
| 
|
| Concentration | Amount of drug in a given volume of plasma. | 83.3 mg/L | 
| 
|
| Elimination half-life | The time required for the concentration of the drug to reach half of its original value. | 12 h | 
| 
|
| Elimination rate constant | The rate at which a drug is removed from the body. | 0.0578 h[SUP]−1[/SUP] | 
| 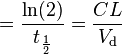
|
| Infusion rate | Rate of infusion required to balance elimination. | 50 mg/h | 
| 
|
| Area under the curve | The integral of the concentration-time curve (after a single dose or in steady state). | 1,320 mg/L·h | 
| 
|
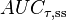
| 
| |||
| Clearance | The volume of plasma cleared of the drug per unit time. | 0.38 L/h | 
| 
|
| Bioavailability | The systemically available fraction of a drug. | 0.8 | 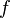
| 
|
| Fluctuation | Peak trough fluctuation within one dosing interval at steady state | 41.8 % | 
| 
where 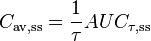
|
<small class="editlink noprint plainlinksneverexpand;">[</small>
<small class="editlink noprint plainlinksneverexpand;">]</small> |
<tbody>
</tbody>
The following graph depicts a typical time course of drug plasma concentration and illustrates main pharmacokinetic metrics:

The time course of drug plasma concentrations over 96 hours following oral administrations every 24 hours. Note that the AUC in steady state equals AUC[SUB]∞[/SUB] after the first dose.
[h=2]Pharmacokinetic models[edit][/h]Pharmacokinetic modelling is performed by noncompartmental or compartmental methods. Noncompartmental methods estimate the exposure to a drug by estimating the area under the curve of a concentration-time graph. Compartmental methods estimate the concentration-time graph using kinetic models. Noncompartmental methods are often more versatile in that they do not assume any specific compartmental model and produce accurate results also acceptable for bioequivalence studies. The final outcome of the transformations that a drug undergoes in an organism and the rules that determine this fate depend on a number of interrelated factors. A number of functional models have been developed in order to simplify the study of pharmacokinetics. These models are based on a consideration of an organism as a number of related compartments. The simplest idea is to think of an organism as only one homogenous compartment. This monocompartimental model presupposes that blood plasma concentrations of the drug are a true reflection of the drug’s concentration in other fluids or tissues and that the elimination of the drug is directly proportional to the drug’s concentration in the organism (first order kinetics).

Graph representing the monocompartmental action model.
However, these models do not always truly reflect the real situation within an organism. For example, not all body tissues have the same blood supply, so the distribution of the drug will be slower in these tissues than in others with a better blood supply. In addition, there are some tissues (such as the brain tissue) that present a real barrier to the distribution of drugs, they can be breached with greater or lesser ease depending on the drug’s characteristics. If these relative conditions for the different tissue types are considered along with the rate of elimination, the organism can be considered to be acting like two compartments: one that we can call thecentral compartment that has a more rapid distribution, comprising organs and systems with a well-developed blood supply; and a peripheral compartment made up of organs with a lower blood flow. Other tissues, such as the brain, can occupy a variable position depending on a drug’s ability to cross the barrier that separates the organ from the blood supply.
This two compartment model will vary depending on which compartment elimination occurs in. The most common situation is that elimination occurs in the central compartment as the liver and kidneys are organs with a good blood supply. However, in some situations it may be that elimination occurs in the peripheral compartment or even in both. This can mean that there are three possible variations in the two compartment model, which still do not cover all possibilities.[SUP][6][/SUP]
This model may not be applicable in situations where some of the enzymes responsible for metabolizing the drug become saturated, or where an active elimination mechanism is present that is independent of the drug's plasma concentration. In the real world each tissue will have its own distribution characteristics and none of them will be strictly linear. If we label the drug’s volume of distribution within the organism Vd[SUB]F[/SUB] and its volume of distribution in a tissue Vd[SUB]T[/SUB] the former will be described by an equation that takes into account all the tissues that act in different ways, that is:
<center style="color: rgb(37, 37, 37); font-family: sans-serif; font-size: 14px; line-height: 22.4px;">
[h=3]Noncompartmental analysis[edit][/h]Noncompartmental PK analysis is highly dependent on estimation of total drug exposure. Total drug exposure is most often estimated by area under the curve (AUC) methods, with the trapezoidal rule (numerical integration) the most common method. Due to the dependence on the length of 'x' in the trapezoidal rule, the area estimation is highly dependent on the blood/plasma sampling schedule. That is, the closer time points are, the closer the trapezoids reflect the actual shape of the concentration-time curve.
[h=3]Compartmental analysis[edit][/h]Compartmental PK analysis uses kinetic models to describe and predict the concentration-time curve. PK compartmental models are often similar to kinetic models used in other scientific disciplines such as chemical kinetics and thermodynamics. The advantage of compartmental over some noncompartmental analyses is the ability to predict the concentration at any time. The disadvantage is the difficulty in developing and validating the proper model. Compartment-free modelling based on curve stripping does not suffer this limitation. The simplest PK compartmental model is the one-compartmental PK model with IV bolus administration and first-order elimination. The most complex PK models (called PBPK models) rely on the use of physiological information to ease development and validation.
[h=3]Single-compartment model[edit][/h]Linear pharmacokinetics is so-called because the graph of the relationship between the various factors involved (dose, blood plasma concentrations, elimination, etc.) gives a straight line or an approximation to one. For drugs to be effective they need to be able to move rapidly from blood plasma to other body fluids and tissues.
The change in concentration over time can be expressed as C=C[SUB]initial[/SUB]*E^(-kelt)
[h=3]Multi-compartmental models[edit][/h]

Graphs for absorption and elimination for a non-linear pharmacokinetic model.
The graph for the non-linear relationship between the various factors is represented by a curve, the relationships between the factors can then be found by calculating the dimensions of different areas under the curve. The models used in 'non linear pharmacokinetics are largely based on Michaelis-Menten kinetics. A reaction’s factors of non linearity include the following:
- Multiphasic absorption : Drugs injected intravenously are removed from the plasma through two primary mechanisms: (1) Distribution to body tissues and (2) metabolism + excretion of the drugs. The resulting decrease of the drug's plasma concentration follows a biphasic pattern (see figure).

Plasma drug concentration vs time after an IV dose
- Alpha phase: An initial phase of rapid decrease in plasma concentration. The decrease is primarily attributed to drug distribution from the central compartment (circulation) into the peripheral compartments (body tissues). This phase ends when a pseudo-equilibrium of drug concentration is established between the central and peripheral compartments.
- Beta phase: A phase of gradual decrease in plasma concentration after the alpha phase. The decrease is primarily attributed to drug metabolism and excretion.[SUP][7][/SUP]
- Additional phases (gamma, delta, etc.) are sometimes seen.[SUP][8][/SUP]
- A drug’s characteristics make a clear distinction between tissues with high and low blood flow.
- Enzymatic saturation: When the dose of a drug whose elimination depends on biotransformation is increased above a certain threshold the enzymes responsible for its metabolism become saturated. The drug’s plasma concentration will then increase disproportionately and its elimination will no longer be constant.
- Induction or enzymatic inhibition: Some drugs have the capacity to inhibit or stimulate their own metabolism, in negative or positive feedback reactions. As occurs with fluvoxamine, fluoxetine and phenytoin. As larger doses of these pharmaceuticals are administered the plasma concentrations of the unmetabolized drug increases and the elimination half-life increases. It is therefore necessary to adjust the dose or other treatment parameters when a high dosage is required.
- The kidneys can also establish active elimination mechanisms for some drugs, independent of plasma concentrations.

[h=2]Bioavailability[edit][/h]Main article: Bioavailability
At a practical level, a drug’s bioavailability can be defined as the proportion of the drug that reaches its site of action. From this perspective the intravenous administration of a drug provides the greatest possible bioavailability, and this method is considered to yield a bioavailability of 1 (or 100%). Bioavailability of other delivery methods is compared with that of intravenous injection («absolute bioavailability») or to a standard value related to other delivery methods in a particular study («relative bioavailability»).
<center style="color: rgb(37, 37, 37); font-family: sans-serif; font-size: 14px; line-height: 22.4px;">
Therefore, if a drug has a bioavailability of 0.8 (or 80%) and it is administered in a dose of 100 mg, the equation will demonstrate the following:
<center style="color: rgb(37, 37, 37); font-family: sans-serif; font-size: 14px; line-height: 22.4px;">De = 0.8 x 100 mg = 80 mg</center>That is the 100 mg administered represents a blood plasma concentration of 80 mg that has the capacity to have a pharmaceutical eff
This concept depends on a series of factors inherent to each drug, such as:[SUP][9][/SUP]
- Pharmaceutical form
- Chemical form
- Route of administration
- Stability
- Metabolism